Introduction
Lipoprotein(a) (Lp(a)) is a complex particle, which acts as a natural blood thinner by linking to plasminogen. It was described by Berg in 1963 for the first time [1]. Lp(a) has many common features with low-density lipoprotein cholesterol (LDL-C), although its levels of distribution in the plasma is skewed reaching levels > 387.8 nmol/l [2]. The function of Lp(a) is not yet fully understood but it is known that there is an interaction between Lp(a) and other established and potential cardiovascular risk factors, such as LDL-C, high-density lipoprotein cholesterol (HDL-C), and homocysteine [3]. Genetic, large-scale, and prospective cohort studies have indicated that high plasma concentrations of Lp(a) increase the risk of CVD and stroke [4–7]. Increased concentrations of Lp(a) have been reported to enhance the risk of death from vascular events and stroke in elderly men independent of other risk factors, including LDL-C [8]. Studies on humans have revealed the effects of Lp(a) in thrombosis, foam cell formation, and inflammation of intima, which are all related to atherosclerosis [9–14]. Patients with lipoprotein(a) > 50 mg/dl but with a low number or ASCVD risk factors (e.g. smoking, diabetes, hypertension, unhealthy diet) had only one third of ASCVD risk for the subsequent 11.5 years compared to those with a high number of risk factors [15]. Therefore, several pharmacological and non-pharmacological options have been investigated as potential modulators of Lp(a) concentrations [16–24], and novel approaches are being developed to reduce this causal risk factor for atherosclerotic cardiovascular disease [25–28]. However, most of the explored agents appear to exert weak Lp(a)-lowering effects considering the findings of observational and Mendelian randomization studies which imply a reduction of around 50–100 mg/dl (based on primary or secondary prevention setting) would be necessary to achieve an atherosclerotic CVD benefit [29, 30].
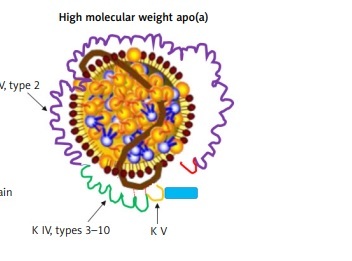
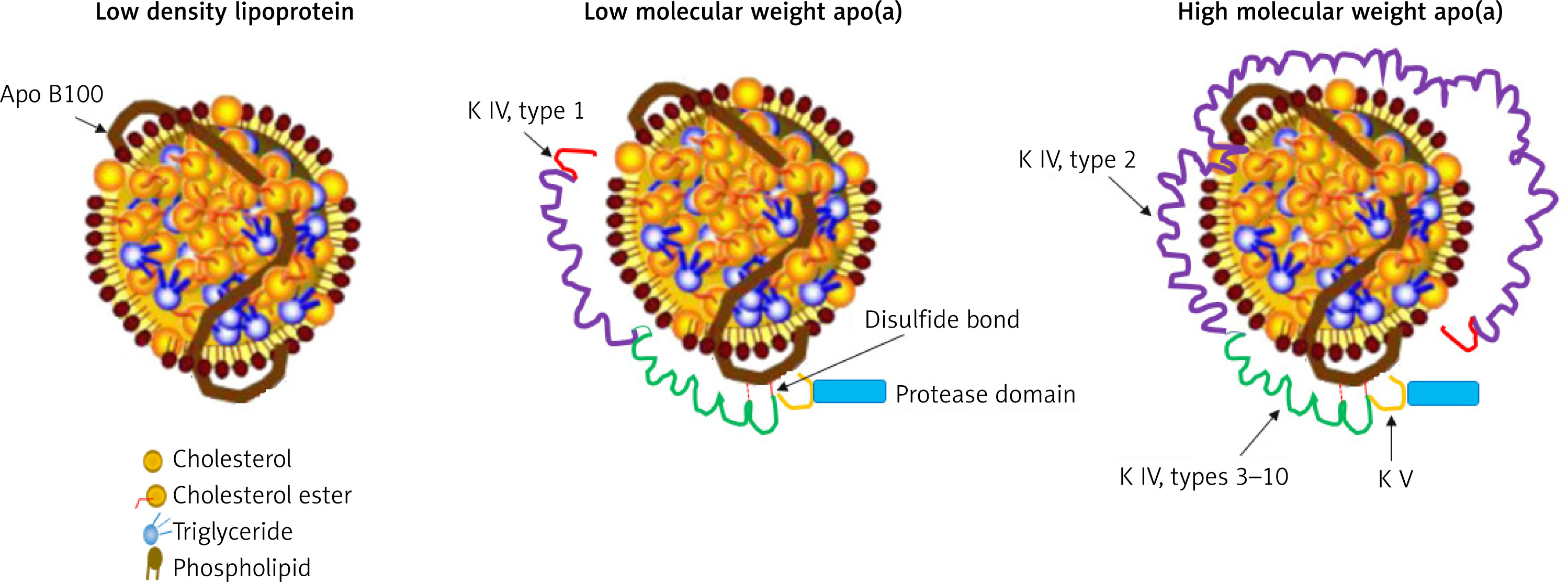
Lp(a) is composed of a cholesterol-rich LDL particle, containing apolipoprotein B-100 and another protein, apo(a), attached by a disulfide bridge (Figure 1) [31, 32]. Apo(a) is a complex protein sharing a high sequence homology with several regions of plasminogen, including the protease domain, and the so-called kringle IV (KIV) and V domains [33]. The amino acid sequence of apo(a) has considerable structural homology with plasminogen. In particular, apo(a) has a kringle structure, which is composed of 10 subtypes (KIV1 to KIV10) and an inactive protease domain. The KIV2 subtype structure is present in multiple repeated copies (1 to > 40 copies) [34]. Apo(a) concentration is generally inversely correlated with apo(a) size and varies widely between individuals. As a consequence of the increased size heterogeneity of apo(a), and of the variable numbers of KIV2 subtypes, the suitable measurement of Lp(a) becomes a real challenge. Moreover, it should be considered that each individual inherits and generally expresses two copies of LPA genes, one from each parent. Thus, unless homozygous for two LPA genes of identical KIV-2 repeat number, most people have two different isoforms of apo(a) and the levels are the sum of the two different apo(a) isoform sizes [35]. These issues are one of the reasons why besides being a CV risk factor, Lp(a) is not routinely evaluated in clinic.
As plasma concentrations of Lp(a) are predominantly genetically determined, they are relatively stable over a lifetime. Therefore, Lp(a) may only need to be measured once, unless a secondary cause is suspected or a specific treatment is instituted in order to lower its plasma concentration (Table I) [36]. Overall, Lp(a) could be measured in all cases of coronary disease, premature CVD, a family history of hypercholesterolemia and premature CVD, or a family history of high Lp(a) [37, 38]. Another aspect worth of consideration is the risk threshold. Lp(a) levels vary greatly between individuals of different ethnicity, with higher plasma Lp(a) levels detected in South Asians compared with Chinese and non-Hispanic whites; Lp(a) concentrations are 2- to 3-fold higher in blacks compared with whites [39]. Ethnic variability in plasma Lp(a) levels implies the need for defining population-specific Lp(a) thresholds for assessing CV risk [40]. However, data from the UK Biobank showed that regardless of ethnicity there is no a threshold at which the risk starts to increase but there is a continuous scale [7].
Table I
Cutoffs reported by the major international clinical practice guidelines
| AHA/ACC 2018 [87] | EAS/ESC 2019 [88] | HEART-UK 2019 [89] | NLA 2019 [90] | Canadian Cardiovascular Society 2021 [91] |
|---|---|---|---|---|
| Lp(a) ≥ 50 mg/dl or ≥ 125 nmol/l may be considered a risk-enhancing factor | Lp(a) ≥ 180 mg/dl (≥ 430 nmol/l) risk equivalent to that associated with HeFH | Minor: 32–90 nmol/l Moderate: 90–200 nmol/l High: 200–400 nmol/l Very high > 400 nmol/l | ≥ 50 mg/dl or ≥ 100 nmol/l | 50 mg/dl (or 100 nmol/l) |
Given the important prognostic roles of Lp(a) levels in CVD risk stratification, accurate determination of this lipoprotein is of utmost importance. Indeed, all the recent evidence clearly showed that Lp(a) should be measured without ifs and buts [41]. Most importantly, the addition of Lp(a) to ASCVD algorithms leads to a net reclassification of the risk, e.g., 31% in the case of SCORE [42].
Several methods have been used for the measurement of Lp(a) in human plasma or serum samples, including radial immunodiffusion (RID) [32], electroimmunoassay [43], radioimmunoassay (RIA) [44], enzyme-linked immunosorbent assay (ELISA) [45–49], immunoturbidimetry [50], nephelometry [51], dissociation-enhanced lanthanide fluorescent immunoassay (DELFIA) [52], particle concentration fluorescence immunoassay (PCFIA) [53], and electrophoretic [54–56] and immunofixation electrophoresis [57]. In order to overcome shortcomings related to commercially available immunoassays, in 2000 a reference material was chosen to be used by manufacturers of commercially available methods to assign a secondary accuracy-based Lp(a) target value to their assay calibrators. This reference material is formed by a pool of serum obtained by the blood of 17 donors. A target value of 107 nmol/l was assigned to the proposed reference material [58]. Table II lists the advantages and disadvantages of reported assays.
Table II
Strengths and limitations of various Lp(a) measuring methods
Lp(a) concentrations have commonly been expressed as the total mass of major components of lipoprotein in mg/dl, although this approach has significant limitations that cannot demonstrate potential variables in the components. However, reporting the number of total apo(a) particles can address this problem. Importantly, a method that can measure the heterogeneity of apo(a) must be used, since this is an important source of variation in the assays, and thus, needs to be addressed for the successful development of a standardized test.
Overview of detection methods
ELISA
ELISA is a plate-based assay technique used for analyzing antibodies, peptides, proteins, and hormones. Several ELISA methods for Lp(a) measurement have been used. An ELISA for Lp(a) determination in plasma was developed by Duvic et al. using a mouse monoclonal antibody, LHLP-1. By using this method, the within-day and between-day variation was about 8% and 12%, respectively. Additionally, the logarithm of the Lp(a) concentration was linear from about 11 to 1408 ng/ml [59].
A sandwich-based, non-competitive ELISA was described by Abe et al. for the determination of Lp(a). In this assay, POD-Fab′ (anti-Lp(a) antibody was used as an enzyme-conjugated antibody to enhance sensitivity. This assay is able to measure Lp(a) over the range of 0.5 to 50 ng/well, with a LOD of 0.5 ng/well [60].
Another ELISA technique was developed for the measurement of Lp(a) concentrations in baboons. The assay has been documented to quantify Lp(a) concentrations from l to 9 ng [61].
In a closely-related approach, an ELISA technique was reported for the detection of apolipoprotein(a) in human Lp(a) in serum using five mouse monoclonal antibodies. This method allowed a wide linear detection range for Lp(a) from 1 to 500 ng/ml and a LOD of 31 ±6.2 ng [62].
The sandwich-based ELISA methods described above utilized a monoclonal antibody as an anti-apo(a) and a polyclonal antibody against apo B, connected with peroxidase, for the detection of Lp(a). The ELISA methods described exhibited a wide linear range for analysis of Lp(a) from 10 to 1000 mg/l [63].
A selective bi-site ELISA assay has also been developed for the detection of Lp(a) using monoclonal antibodies that is based on the conjugation of apo(a) to apoB. This immunochemical assay allows for Lp(a) detection within the linear working range of 0.06–0.40 µg/ml [64].
Fless et al. developed a sandwich ELISA for plasma Lp(a) using anti-apo(a) as the capture antibody. In this particular assay, the presence of plasminogen caused insignificant interference with the detection of Lp(a). Using this method, Lp(a) was determined within a linear range of 0.045 to 13.3 mg/dl, with a LOD of 0.030 mg/dl [48].
Another ELISA-based method for the detection of Lp(a) was described using a monoclonal antibody. The assay allowed for Lp(a) measurements over a linear range from 0.5 to 180 ng/ml [65].
An indirect sandwich ELISA was designed for quantitation of Lp(a) in both serum and dried blood spots using commercially-available materials. Sheep polyclonal anti-human Lp(a) antiserum and rabbit anti-human Lp(a) polyclonal antiserum were used as the ‘capture antibody’ and the ‘detection antibody’, respectively. Goat anti-rabbit IgG-HRPO (horseradish peroxidase) conjugate was the labeled secondary antibody. The calibration curve demonstrated a linear range from 5 to 160 µg/l, with a LOD of less than 5 µg/l. Furthermore, these authors evaluated the correlation between Lp(a) concentrations measured by ELISA and by RIA. They reported a large difference between these two methods (34.3 ±9.7%) [47].
Another ELISA-based method was developed by Morikawa et al. They reported a competitive two-step monoclonal ELISA method for the measurement of Lp(a) in serum. Using this method, a linear dynamic range from 2 to 1000 mg/l with a LOD of 2 mg/l was achieved [46].
Marcovina et al. developed three direct-binding ELISA assays using the same monoclonal antibodies to investigate the effect of apo(a) size polymorphism on the immunochemical quantification of Lp(a) [49]. The one developed at Northwest Lipid Metabolism and Diabetes Research Laboratories is and ELISA isoform independent giving values in nmol/l. It is considered the gold standard. In order to capture the apo(a) particles, wells are coated with the murine monoclonal antibody a-6 directed to apo(a) KIV2 repeats, whereas the detection is by means of the murine monoclonal antibody a-40 which is direct towards a single epitope present in apo(a) KIV9 [66]. Although this assay recognized apo(a) free in plasma, this percentage is less than 5% and thus negligible in terms of interpretation of the assay [67].
An in-house ELISA assay has been also developed by the University of California, San Diego (UCSD) research laboratory. The assay captures all the apoB-100–containing lipoprotein particles by meaning of murine monoclonal antibody MB4721 and detecting apo(a) with biotinylated murine monoclonal antibody LPA4. This latter binds to the epitope NYCRNPDA which is also present in the KIV2 repeats. This assay is, therefore, affected by the apo(a) isoform polymorphism and reports the values in mg/dl of total Lp(a) mass [68].
Finally, a sandwich ELISA method for apo(a) in serum samples has been described by Yamada et al. in which the size heterogeneity of apo(a) did not affect the assay results. The reported recovery of this assay was 97 to 105%. The working range for detection of this technique was 1.5–280 nmol/l with a LOD of 1.5 nmol/l [45]. In general, these reported assay methods have many limitations, such as a lack of similar specificity, accuracy, and sensitivity for the different isoforms of Lp(a), in addition to the systems being fairly complicated.
In conclusion, these assays use an antibody against apo(a) that does not recognize a unique epitope in each particle and cross-react with multiple KIV-2 domains. This could lead Lp(a) levels to be overestimated or underestimated in clinical samples containing large or small isoforms, respectively [67]. So far, the Denka-based assay is the least isoform sensitive, mainly due to the use of five calibrators to cover the measured range of concentrations, each calibrator being independent and containing a suitable distribution of apo(a) isoforms, traceable in nmol/l: the high-level calibrators contain small isoforms and the low-level calibrators containing large ones [69].
Fluorescence
Fluorescent immunoassays are immunoreagents labeled directly with fluorescent compounds, or their precursors [70]. This type of assay is based on a biochemical technique that monitors the binding of the “detection” antibody and the target molecule. This technique has many advantages, such as speed, high sensitivity, simplified reagents, and a relatively simple assay design. Therefore, fluorescent immunoassays have been used to measure Lp(a).
Jurgens et al. developed a dissociation-enhanced lanthanide fluorescence method based on a solid-phase immunoassay for the detection of Lp(a) in serum. A polyclonal antiserum was used against apo-B, and the polyclonal antiserum was also used against Lp(a). This system provided a linear quantification of Lp(a) for anti-apo-B and anti-Lp(a) over a wide range; specifically, up to 1800 mg/l and 1900 mg/l, with LOD values of 4 mg/l and 2.5 mg/l, respectively [52].
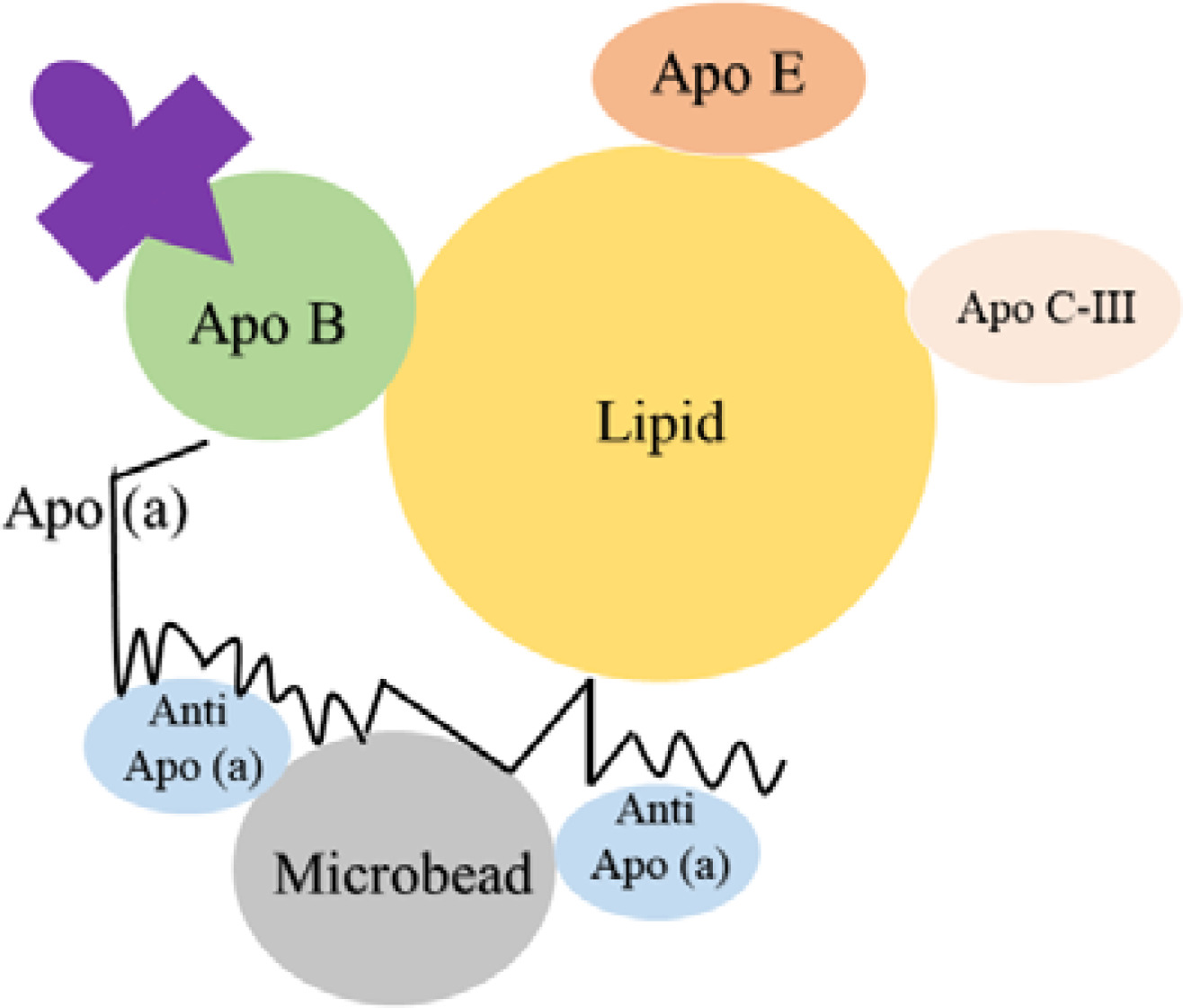
A particle concentration fluorescence immunoassay (PCFIA) for Lp(a) was developed by Kottke et al., which utilized a mixture of monoclonal antibodies specific for apo(a). These antibodies are bound to inert microscopic beads to capture the Lp(a) particles. Subsequently, a monoclonal antibody labeled with fluorescein against apo-B is used for the detection and quantitation as shown in Figure 2. This method relied on apo-B, so it was independent of variations in the size of apo(a). By using PCFIA, the Lp(a) level was detected over a wide range (from 4.65 to 63.87 mg/dl), with negligible interference from plasminogen and apo-B [53].
The major limitation of fluorescence methods discussed in this review is that they only measure the apo-B in the Lp(a), and are unable to detect any size variation in apo(a).
Nephelometric and immunoturbidimetric
Immunoturbidimetry and nephelometry both determine the turbidity of a sample to measure the concentration of an analyte. Turbidimetry measures the intensity of light transmitted through the sample, while nephelometry measures the intensity of the scattered light at a fixed angle. Nephelometric and immunoturbidimetric methods have been used to assay Lp(a). Cazzolato et al. used nephelometry for the determination of Lp(a) over a working range of 0.1 to 1.5 g/l [71]. Another nephelometric immunoassay has also been developed by Borque et al. for Lp(a) measurement, which is based on carboxylated latex particles coated with F(ab′)2 fragments in the serum. This assay allows the determination of Lp(a) levels in the linear range from 27 to 1750 mg/l [51].
An immunoturbidimetric assay has been developed to quantify the Lp(a) levels in serum samples. In this assay, the results significantly improved with L-proline and the recovery obtained was 106% (range: 90–116%). This method permitted the quantification of Lp(a) in the linear range from 50 to 1100 mg/l [50].
These two methods described above determined the mass of Lp(a), which is not able to determine apo(a) size and cannot assess size variability of the Lp(a).
Radial immunodiffusion and radioimmunoassay
The easiest available immunoassay method is radial immunodiffusion (RID) or the Mancini method. RID is based on the classic precipitin reaction of antigen and antibodies, in which the reaction forms a precipitate and, thus, directly determines the concentration of IgG in serum or plasma. RID has previously been used for the detection of Lp(a) in plasma. Albers et al. developed an immunochemical assay for Lp(a) in plasma using RID. The assay permitted a sensitive measurement of Lp(a) at concentrations above 8 mg/dl, with a lower limit of sensitivity of 1.5 mg/dl [32].
Another RID assay was developed for the measurement of Lp(a) in human plasma. When used to determine the concentration of Lp(a) lipoprotein in the plasma of 27 Lp(a+) individuals, values ranged from 84 mg/dl to less than 2.8 mg/dl, with a mean of 21.3 mg/dl [72].
To enhance the selectivity and sensitivity of Lp(a) analysis, a double-antibody radioimmunoassay was developed. The between-assay coefficient of variation of this technique was very low (8%) [44].
It should be noted that radioimmunoassay methods have limitations, such as high coefficients of variation and low sensitivity. This method is also more time consuming and complex than other immunoassays. In addition to these limitations, the major shortcoming with this method is the fact that it is impossible to determine the size variability of Lp(a).
Electrophoresis methods
Electrophoresis is a separation technique that is based on the migration of charged particles (ions) under the influence of an electric field [73]. Electrophoresis has been used as a routine assay in many clinical laboratories for the measurement of serum lipoproteins and Lp(a) levels. For electrophoresis of serum lipoproteins, numerous supporting media must be utilized, for example, agarose [74, 75] and cellulose acetate [75].
Immunofixation electrophoresis
Immunofixation electrophoresis is the most sensitive method for the detection and ‘typing’ of monoclonal antibodies or immunoglobulins. The specimen to be analyzed can be urine, serum, or other body fluids. A lipoprotein immunofixation electrophoresis (Lipo-IFE) for measuring the concentration of Lp(a) particles (Lp(a)-P) was developed by Guadagno et al. using polyclonal apo-B antibodies. The method enabled the analysis of Lp(a) in a linear range from 50 to 800 nmol/l, with a LOD of 20 nmol/l [57]. This method measured Lp(a)-P concentration (nmol/l). By using the aforementioned method, it was possible to quantify apo-B, but the size of apo(a) could not be measured, which was the major limitation of this method.
Counterimmunoelectrophoresis
Counterimmunoelectrophoresis (CIEP) has been used to detect Lp(a). This technique is based on immunoprecipitation that utilizes electrophoresis to increase the rate of migration of the antigen and antibodies in a gel matrix [76]. Molinari et al. developed a useful method based on CIEP for screening serum for pathological Lp(a) value greater than 0.3 g/l. This method achieved a detection limit of 0.285 g/l without considerable interference from triglyceride and cholesterol [77]. Historically, CIEP has been more sensitive than conventional double immunodiffusion, but the main limitation associated with this technique is the inability to assay apo(a).
Electroimmunoassay (EIA) or rocket immunoelectrophoresis
Immunoelectrophoresis is a technique that is used for the separation and determination of proteins based on differences in electrical charge and their reactivity with antibodies. Rocket immunoelectrophoresis is a one-dimensional immunoelectrophoresis that has been used for the measurement of human serum proteins. An EIA was reported by Strobl et al. for the determination of Lp(a) in cord and capillary serum. Within-run coefficients of variation for this method ranged from 2% to 5%. The mean levels of Lp(a) in cord serum were 3.1–4.4 mg/dl and the mean value of Lp(a) in the serum of adults was 15 mg/dl [78].
Guyton et al. also quantified plasma Lp(a) levels using an EIA method. These authors reported a coefficient of variation of 4.2% and a mean plasma Lp(a) concentration of 16.3 mg/dl [79]. In yet another approach to the analysis of Lp(a), Gries et al. investigated the binding of various monoclonal antibodies (Mabs) (2A, 9A, 6B, L3, L7) against apo-B to Lp(a), LDL, and reduced Lp(a). All of the Mabs showed a suitable affinity to apo-B for the different lipoproteins evaluated [43].
In a closely-related approach, Marz et al. analyzed serum Lp(a) by zone immunoelectrophoresis (ZIA), and compared it to results obtained using EIA. The two methods for the determination of serum Lp(a) showed a good correlation with each other. The resultant coefficients of variation for the ZIA were estimated to be 12% for inter-assay variation. ZIA and EIA exhibited linear quantification of Lp(a) from 3 to 40 mg/l and 10 to 100 mg/l, respectively [80]. However, an important disadvantage of the immunoelectrophoresis method was the inability to measure Lp(a) heterogeneity.
Laurell electrophoresis (rocket electrophoresis)
Rocket electrophoresis is another technique for determining the concentration of a specific protein in a protein mixture. This method, which was initially developed by Laurell, is also known as electroimmunoassay or electroimmunodiffusion [81]. Laurell electrophoresis has previously been utilized for the analysis of Lp(a) concentrations in serum. This technique is sensitive in the range of 1 to 60 mg/dl with a day-to-day coefficient of variation of less than 4% [55, 56].
Finally, Kawakami et al. has reported a rapid electrophoretic method using an agarose gel film for the measurement of serum Lp(a). This method was able to discriminate between Lp(a-) (Lp(a)-negative) and Lp(a+) (Lp(a)-positive) in human subjects [54]. In fact, this method measures Lp(a) qualitatively.
Lastly, it is worth noting that many laboratories use these routine electrophoretic techniques for the determination of Lp(a) in serum. However, these methods cannot measure the size variability associated with Lp(a), which is the main limitation of these techniques.
From a historical perspective, the first commercial kits determined Lp(a) by use of radioimmunoassay or radial immunodiffusion [44]. Current commercially-available immunoassays for Lp(a) measurement are ELISA, immunoturbidometric, and immunonephelometric assays that use specific antibodies against the apo(a) moiety of Lp(a) [82]. Although the International Federation of Clinical Chemistry (IFCC) has expended much effort to develop a reference material for the standardization of the analytical methods, commercially-available immunoassays for such complex particles have many shortcomings. A lack of sensitivity is the primary shortcoming due to variance in the size of apo(a) isoforms. The current lack of universal (reference) standards when measuring Lp(a) is also another problem [83].
Liquid chromatography–mass spectrometry
Considering that targeted liquid chromatography tandem mass spectrometry (LC-MS/MS) has become a method of choice for the standardization of biomarkers in clinical practice, it has also been tested in the case of Lp(a) evaluation [84]. Among others, the add-on value of LC-MS/MS is that it is traceable to SI units through a calibration strategy such as isotope dilution [85]. A parallelism between gold standard ELISA and LC-MS/MS was performed on a set of 64 samples with well-characterized apo(a) isoforms. Results obtained by the LC-MS/MS method and those obtained by the gold standard ELISA yielded y = 0.98 × ELISA + 3.18. A further validation of this approach was the excellent agreement with the value of the secondary reference material WHO/IFCC SRM-2B, namely, it was 104.7 ±8.4 nmol/l compared to the assigned value of 107 nmol/l [85].
Conclusion and future perspective
The measurement of Lp(a) is surprisingly complex. Although several immunoassays, fluorescence-based assays, and electrophoretic methods have been developed for Lp(a), standardization of the assay for Lp(a) has been impossible due to different antibody reactivities to various Lp(a) phenotypes. The strengths and limitations of various methods used to measure Lp(a) are summarized in Table II. Considering that to convert the values from mg/dL to nmol/L or vice versa is not a straightforward process and it is to avoid/to be avoided, to develop a candidate reference method for the standardization of analytical methods to measure Lp(a) in nmol/l is important in recognizing those individuals that may be at risk for CVD. As presented in Table III, no assay has been reported to be 100% insensitive to apo(a) size. However, it has to be recognized that those assays using 5 independent calibrators with a large range of Lp(a) levels and a suitable distribution of apo(a) isoforms have reduced the impact of apo(a) size [86]. Thus, there is an urgent need to cover a gap in the existing knowledge as it pertains to the complete measurement of Lp(a) since the major limitation associated with existing methods is their inability to measure the variability/heterogeneity in Lp(a) size. With this limitation in mind, it is recommended to use methods that determine the size of proteins (nmol/l), such as dynamic light scattering (DLS), multi-angle light scattering analysis, near-field imaging, sedimentation, gel filtration, and electron microscopy. Therefore, we would suggest that these methods/assays hold promise in resolving the limitations associated with the measurement of Lp(a) in biological matrices.
Table III
Comparison between reported assays for measuring Lp(a)
| Analytical method | Sample | Detection limit | Working range | CV% | Mean | SD | Recovery (%) | Throughput | Time | Interference | Ref. |
|---|---|---|---|---|---|---|---|---|---|---|---|
| ELISA | Plasma | – | 11 to 1408 ng/ml | 8% and 12% | – | 0.07% | 86.3 to 97.1% | – | – | – | [59] |
| ELISA | Serum and cord blood | 0.5 ng/well | 0.5 to 50 ng/well | 3.5% to 11.1% | 4.9 to 103.8 | 0.51 to 2.41 | 102.3 ±4.6 to 105.7 ±3.7 | Chylomicrons, hemoglobin, bilirubin | [60] | ||
| ELISA | Baboon sera | – | l to 9 ng | 8% to 9 % | 4.7 mg/dl | – | – | Serum proteins | [61] | ||
| ELISA | Serum | 31 ±6.2 ng | 1 to 500 ng/ml | – | – | – | – | – | – | Plasminogen, LDL-apoB-100 and other apolipoproteins | [62] |
| ELISA | Plasma | – | 10 to 1000 mg/l | 3.0% to 5.6% | 120 mg/l | 130 mg/l | – | HDL cholesterol, apo AI, ape B, triglyceride, apo AII and plasma cholesterol | [63] | ||
| ELISA | Plasma | – | 0.06 to 0.40 µg/ml | 4.7% to 9.6% | – | – | 85% to 100% | Plasminogen | [64] | ||
| ELISA | Plasma | 0.030 mg/dl | 0.045 to 13.3 mg/dl | 3.2 mg/dl | – | – | Plasminogen | [48] | |||
| ELISA | Plasma | – | 0.5 to 180 ng/ml | 2.8% to 10.7% | 13.8 to 299.5 mg/l | 1.4 to 9.8 mg/l | 88.3 – 117.0% | Plasminogen | [65] | ||
| ELISA | Serum and dried blood | < 5 µg/l | 5 to 160 µg/l | 6.3 ±2.3% and 4.5 ±0.1% | 300 mg/l | 36.3–50.2% | – | – | – | – | [47] |
| ELISA | Serum | 2 mg/l | 2 to 1000 mg/l | 2.9–3.9% and 3.4–4.0% | 96.4 to 112.6 mg/l | 24.7 to 25.7 mg/l | 93.5 to 103.1% | Plasminogen and LDL | [46] | ||
| ELISA | Serum | – | ≈0.25 to 4.75 nmol/l | ≤ 10% | – | – | – | – | – | – | [49] |
| ELISA | Serum | 1.5 nmol/l | 1.5 to 280 nmol/l | 1.5–7.5 % | – | – | 97 to 105% | – | – | LDL | [45] |
| Fluorescence | Serum | 2.5 mg/l | Up to 1900 mg/l | 2.7 to 15% | 81 to 1103 mg/l | 5.8 to 76 mg/l | 96.75 to 103.4% | Plasminogen | [52] | ||
| Fluorescence | Plasma | – | 4.65 to 63.87 mg/dl | 5.36 to 18.05% | 4.45 to 70.632 | 0.239 to 10.96 | – | Pplasminogen and apo B | [53] | ||
| Nephelometric | Serum | – | 0.1 to 1.5 g/l | 1.2 to 3.9% | – | 0.253 to 0.274 | – | – | – | – | [71] |
| Nephelometric | Serum | – | 27 to 1750 mg/l | 2.9 to 7.8% | 52 to 534 | 1.9 to 16.3 | 96.5 to 108.76% | – | – | Hemoglobin, bilirubin, intralipid, plasminogen, apo B and rheumatoid | [51] |
| Immunoturbidimetric | Serum | – | EI50 to 1100 mg/1 | < 10% | 35112 mg/1 | ±2643 | 106% | – | – | Plasminogen and LDL | [50] |
| Electroimmunoassay (EIA) | Cord serum and capillary serum | – | – | 2–5% | In cord serum: 3.1 to 4.4 mg/dl and in serum of adults: 15 mg/dl | 2.5 to 17.5 | – | – | – | – | [78] |
| Electroimmunoassay (EIA) | Plasma | – | –– | 4.2% | 16.3 mg/dl | – | – | – | – | – | [79] |
| Electroimmunoassay (EIA) | Serum | – | – | – | – | – | – | – | – | Apo[a] | [43] |
| Radial immunodiffusion | Plasma | – | Above 8 mg/100 ml | 7 to 27% | 14 mg/100 ml | – | – | – | – | – | [32] |
| Radial immunodiffusion | Plasma | – | 84 to 2.8 mg/100 ml | – | 21.3 mg/100 ml | – | 8.8 – 11.5% | – | – | – | [72] |
| Radioimmunoassay | Plasma | – | > 0.5 mg/dl | 8% | 0.5 mg/dl | 0.1 mg/dl | – | – | – | Apolipoprotein A-I and A-II | [44] |
| Immunofixation electrophoresis | Serum | 20 nmol/l | 50 to 800 nmol/l | < 10% | – | – | – | 100 | 90 min | Bilirubin, hemoglobin and lipemia | [57] |
| counterimmunoelectrophoresis | Serum | 0.285 g/l | 0.3 g /l to 3.35 g/l | – | – | – | – | – | – | Cholesterol, triglycerides | [77] |
| a) Electroimmunoassay, b) Zone immunoelectrophoresis | Serum | – | a) 10 to 100 mg/l b) 3 to 40 mg/l | a) – b) 12% | a) – b) 5 females: 83, males: 136 | a) – b) females: ±54, –males: ±134 | – | – | – | – | [80] |
| Rocket electrophoresis | Serum | – | l to 60 mg/dl | 4% | – | – | – | – | – | – | [55, 56] |
| Electrophoresis | Serum | – | – | – | – | – | – | – | – | – | [54] |