Introduction
Diabetic kidney disease (DKD) (also termed diabetic nephropathy or chronic kidney disease (CKD) due to diabetes) is described in approximately 20–40% of all diabetic patients and is associated with significant cardiovascular and all-cause mortality [1]. Although DKD is typically associated with proteinuria, followed by a progressive kidney decline, a significant percentage of patients with type 2 diabetes (T2D) experience decrease in renal function without proteinuria (known as non-proteinuric DKD); intrarenal atherosclerosis plays a pivotal role in this renal phenotype [2]. Sodium-glucose co-transporter 2 (SGLT2) inhibitors and GLP-1 receptor (GLP-1R) agonists have achieved major cardiovascular and renal benefits in patients with T2D, signifying a new era, in which therapeutic approaches should be tailored based on patients’ comorbidities and on end-organ protection, rather than focusing only on reducing hyperglycaemia [3–6]. In the recent Kidney Disease: Improving Global Outcomes (KDIGO) 2020 guidelines for the management of diabetes in CKD it was stated that metformin (MET) and SGLT2 inhibitors should be the initial combination of choice for most patients with DKD, who have an estimated glomerular filtration rate (eGFR) ≥ 30 ml/min/1.73 m2. GLP-1R agonists were generally preferred as a second line therapeutic approach [7].
Pioglitazone and the kidney
Thiazolidinediones (TZDs) are powerful exogenous agonists of the nuclear peroxisome proliferator activated receptor-γ (PPAR-γ) [8]. They augment the effects of insulin to its cellular targets (especially to the adipose tissue) and are currently the only true sensitizing antidiabetic medications in our therapeutic armamentarium [8–10]. PPAR-γ2 is found almost exclusively in adipose tissue [8]. PPAR-γ1 is widely found in several tissues and has an important role in renal physiology. It is expressed in glomerular, mesangial and tubular segments (especially the renal medullary collecting duct), as well as in renal microvasculature [8, 10]. Pioglitazone (PIO) is currently the main TZD in clinical practice. It is administered in a maximum daily dose of 45 mg for the treatment of T2D [11]. No accumulation of either PIO or its metabolites were found in varying degrees of renal impairment [12].
PIO has achieved remarkable improvements of diabetic albuminuria regardless of baseline kidney function; it was also advantageous in terms of preserving kidney function in uncontrolled T2D patients, as add-on therapy, compared to basal insulin [13–16]. In The Prospective Pioglitazone Clinical Trial In Macrovascular Events (PROactive) study 467 (20.4%) patients in the PIO arm experienced CKD versus 370 (16.3%) patients in the placebo arm (p = 0.0004) [17, 18]. Furthermore, PIO was recently associated with significant improvements in mortality and several other adverse outcomes in patients with T2D and advanced CKD [19]. It also reduced major adverse cardiac events, cerebrovascular events and all-cause mortality, when compared to patients treated with dipeptidyl peptidase-4 (DPP-4) inhibitors, in the same population [20]. In a recent retrospective cohort study organized by Ho et al., PIO administration did not improve any composite hard renal outcomes (sustained eGFR < 15 ml/min/1.73 m2, doubling of serum creatinine levels and the evolution to haemodialysis or kidney transplantation) compared to non-PIO patients, although it was associated with mild nonsignificant albuminuria reduction [21]. However, the relatively short mean follow-up of 2 years, the relatively small sample size and the lack of estimation for medication compliance were the main limitations of this study.
Despite the many beneficial effects of PIO, the controversy for an increased risk of developing bladder cancer, as well as the higher risk for experiencing other side effects (oedema, weight gain, bone fractures and exacerbation of heart failure), have significantly limited its use in routine clinical practice [22, 23]. A large population-based study (2004–2013) of 144,252 older adults with diabetes and CKD (eGFR < 60 ml/min/1.73 m2 or on chronic dialysis) showed that the administration of PIO was very low and gradually declined over time (from 1.9% to 1.7%) [24]. Furthermore, poor awareness and knowledge among medical stuff, for proper dose adjustments of antidiabetic drugs in patients with DKD, was reported to be high; the highest was for PIO, reaching 72.8% [25]. This paper explores the pathophysiological mechanisms governing the possible nephroprotective activity of PIO during a diabetic state. It also discusses its future role in ameliorating the global burden of DKD.
DKD: pathophysiological concepts
Ample evidence suggests that a multifactorial pathogenic process underlies the evolution of DKD; this process has been tightly associated with structural and functional defects [6, 26]. However, hyperglycaemia and its multiple aberrant intracellular and biochemical pathways 9upregulation of the hexosamine and polyol pathway, production of advanced glycation end products (AGEs) and activation of protein kinase C (PKC)) are the major and predominant factors that underlie its pathogenesis; elevated blood pressure (BP) is the next most significant factor [6, 26–28]. After a hyperglycaemic state is established, several pathophysiologic pathways are activated and govern its progression. The most important are: (i) haemodynamic alterations; (ii) tubular factors and hypertrophy; (iii) overactive renin-angiotensin-aldosterone system (RAAS); (iv) increased inflammation, endothelial dysfunction, and oxidative stress; (v) hypoxia; (vi) mitochondrial dysfunction and injury; (vii) podocyte injury and albuminuria; (viii) impaired glomerular autophagy; (ix) activation of sodium-hydrogen exchangers (NHEs); and (x) renal fibrosis [6, 26–28]. The crosstalk among several of these pathways determines the complexity of this disease, while the exact contribution of each pathway to the evolution of DKD is currently under investigation [6, 28]. Obesity or lipotoxicity, insulin resistance (IR) and dyslipidaemia can also stimulate various aberrant cellular mechanisms that promote its development [28]. Glomerulosclerosis and tubulointerstitial scarring are the final cellular pathways. Transforming growth factor β (TGF-β) and its downstream effector connective tissue growth factor (CTGF) are critical hyperplastic and profibrotic cytokines that are involved in this process [6, 28, 29].
Systemic mechanisms of renoprotection
Reduction of hyperglycaemia
Strict glycaemic control is closely related to the reduction of microalbuminuria and protection from renal injury, both in patients with T2D and type 1 diabetes (T1D) [30, 31]. PIO administration has achieved significant and durable reductions of glycated haemoglobin (A1c), reaching 2% (starting with a mean value of 9.3%). Approximately 80% of its efficacy is reached in a daily dose of 30 mg [11, 23]. Inhibition of the hyperglycaemia induced diacylglycerol (DAG)-PKC-extracellular signal-regulated kinase (ERK) aberrant pathway was suggested to play an important role in the restoration of early diabetic glomerular dysfunction, after its administration [32].
Amelioration of IR
IR by itself and/or through subsequent chronic hyperinsulinaemia has been associated with the components of the metabolic syndrome (glucose intolerance, hypertension, dyslipidaemia, hypercoagulability, endothelial and vascular dysfunction); this is described in about 90% of patients with T2D [33]. It has been related to the evolution of albuminuria [34]. It can also cause renal injury through various cellular pathways [34, 35]. Interestingly, insulin sensitivity has been strongly and inversely related to the degree of albuminuria, early during T2D in youth [36].
In vitro data have also suggested that insulin can promote the growth of cultured preglomerular arteriolar smooth muscle cells [37]. It can also stimulate higher renal blood flow rates, since its direct vasodilatory responses to the pre-glomerular (afferent) arterioles are more pronounced compared to those in the post-glomerular (efferent) arterioles, leading to hyperfiltration [38–40]. Increased insulin-induced sodium retention, after enhanced reabsorption in the distal renal tubule, has also been correlated with increased eGFR levels [41, 42].
PIO administration has promoted significant reductions of the homeostasis model assessment of insulin sensitivity (HOMA-S) and fasting insulin levels compared to other oral antidiabetic medications (MET, sulfonylureas (SUs)) [43–46]. It has reduced the progression from oral treatment to insulin therapy in T2D patients. It can also decrease total insulin daily needs by approximately 25% compared to baseline values [13, 47, 48].
BP reduction
TZDs can significantly reduce arterial BP (varying between 10 and 6 mmHg for systolic and diastolic BP respectively); this effect has been associated with significant reduction of the urine albumin-to-creatinine ratio (UACR) [49, 50]. Reduction of BP could be the result of several mechanisms after PIO therapy, namely: (i) direct vasodepressor effect on vascular smooth muscle cells; this effect was achieved after reducing their sensitivity to angiotensin II (Ang II) and by increasing systemic nitric oxide (NO) levels; (ii) improved endothelial function; (iii) attenuation of sympathetic overactivity; (iv) reduction of increased oxidative stress; (v) alterations of circulating and possibly intracellular magnesium levels towards BP reduction; (vi) suppressing calcium entering the cells; (vii) reduction of HOMA-S; (viii) stimulation of genes that belong to the protective axis of the RAAS cascade (Ang II receptor type 2 (AT2R) and Mas receptor pathways), which contribute to vasodilation and apoptosis; and (ix) reduction of renal arteriolar smooth muscle cell proliferation [14, 34, 37, 49, 51]. It was also found to restore normal nocturnal BP declines in non-dippers, parallel to the reduction of the HOMA index, after transforming BP circadian rhythms from a non-dipping to a dipping profile [52].
Improvement of arterial stiffness and endothelial dysfunction
The renal circulatory bed has low vascular resistance, which makes it vulnerable to any possible cellular damage, as pulse wave penetrates deeply into its microvasculature [6, 53]. Arterial stiffness is characterized by increased pulse wave velocity through the arterial vasculature; it promotes increased central systolic blood pressure and elevated central pulse pressure [54]. As aortic stiffness evolves, significant transmission of wider and possibly detrimental pulsatile pressure waves to the renal microcirculation are found [55, 56]. Ample evidence has shown that arterial stiffness is an independent and strong predictor of CKD, while stiffening of the renal microvasculature is closely associated with albuminuria [57–59]. PIO improved endothelial dysfunction and arterial stiffness, both in preclinical diabetic models and in the T2D population (with or without microalbuminuria) through mechanisms beyond its antihyperglycaemic activity [60–62]. Increased adiponectin levels and suppression of nitrative stress have been described as possible key mechanisms that mediate this effect [62–64].
Improvement of diabetic dyslipidaemia
A wealth of evidence suggests that diabetic dyslipidaemia contributes to the evolution of DKD and can be a significant predictor of renal dysfunction [10, 11]. Interestingly, a large population study that analysed data from 47,177 outpatients with T2D (monitored for four years) suggested that low high-density lipoprotein cholesterol (HDL-C) and high triglyceride (TGs) levels were independent risk factors for the development of DKD [65]. PIO has consistently reduced TG levels by approximately 12–30% and exerted a favourable effect on postprandial hypertriglyceridemia [8, 13, 66, 67]. Moreover, in vitro data showed that elevated low-density lipoprotein cholesterol (LDL-C) levels can have a role in the development of tubular dysfunction through increased secretion of proinflammatory and profibrotic cytokines; PIO administration normalized these defects [68].
Direct mechanisms of protection at the kidney level
Renal hyperfiltration and increased intraglomerular pressure
Introduction
Approximately 6% to 73% of patients with T2D experience compensatory hyperfiltration in the whole kidney due to hyperglycaemia and hyperinsulinaemia [6, 69]. The pathogenesis of hyperfiltration and glomerular hypertension is however complex and multifactorial [6, 70]. The two main mechanisms responsible for their evolution are: (i) vascular/neurohormonal factors; and (ii) tubular factors.
The imbalance of vascular/neurohormonal factors results from the disproportion of vasoactive humoral factors that affect the afferent and efferent arteriolar tone (vascular hypothesis). The main molecules that promote a net effect in the reduction of afferent arterial resistance (thereby increasing single-nephron eGFR) are: (i) hyperinsulinaemia per se, (ii) NO bioavailability, (iii) atrial natriuretic peptide (ANP), (iv) Ang (1-7), (v) kallikrein-kinins, and (vi) prostaglandin E2 (PGE2), a major product of cyclooxygenase 2 (COX-2) pathway, which can stimulate renin release. The main molecules that promote a net increase of efferent arterial resistance leading to higher eGFR levels are: (i) endothilin-1 (ET-1), (ii) Ang II, (iii) thromboxane A2 and (iv) reactive oxygen species (ROS) [6, 70].
NO is mainly derived from the neuronal NO synthase (nNOS) in the macula densa and has vital role in the regulation of glomerular blood flow [71]. The two other isoforms which are responsible for its production are inducible NOS (iNOS) and endothelial constitutive NOS (ecNOS). NO can decrease afferent arterial resistance and can promote vasodilation and subsequent hyperfiltration [71, 72]. Other important activities of NO are: (i) mediation of pressure natriuresis; (ii) inhibition of tubular sodium reabsorption; and (iii) modulation of renal sympathetic neural activity [71, 73]. Renal nNOS can affect intrarenal stimulation of the RAAS axis, while ecNOS expression and activity are considered important haemodynamic factors for early glomerular hyperfiltration in diabetes [73].
Activation of tubuloglomerular feedback (TGF) (tubular hypothesis) is the other main mechanism. In diabetic patients SGLT2 upregulation results in maladaptive high-grade glucose and sodium reabsorption in the proximal convoluted tubule (PCT) [6, 28, 70]. Hence distal sodium chloride delivery to the macular densa of the juxtaglomerular apparatus is significantly decreased [6, 74]. This, in turn, results in less intracellular adenosine triphosphate (ATP) breakdown to adenosine. Since adenosine acts as a vasoconstrictor in this area of the nephron, after binding to the adenosine A1 receptors (A1ARs), it promotes an afferent renal vasodilatory response, hyperfiltration and eventually glomerular hypertension [6, 74].
PIO
When PIO was administered in a preclinical model of early-stage diabetic nephropathy, its effect on microalbuminuria was the result of improved hyperfiltration and subsequent glomerular enlargement. The expression of ecNOS was significantly suppressed in the glomerular vessels [75]. In another study organized in Otsuka-Long-Evans-Tokushima Fatty (OLETF) rats, PIO administration restored several parameters related to glomerular hyperfiltration. It improved kidney weight, creatinine clearance, UACR, and glomerular surface; all intensities of nNOS and COX-2 in the macula densa were restored to normal levels [76].
PIO therapy was found to suppress preglomerular arteriole remodelling, when administered in a preclinical model of T2D and obesity, after the reduction of renal homocysteine levels and the suppression of matrix metalloproteinase (MMP)-2 activity [77]. PIO also prevented the evolution of glomerular hyperfiltration through downregulation of the activated DAG-PCK-ERK pathway in mesangial cells [78]. This effect was mainly achieved after the suppression of molecules that cause afferent vasodilation (ANP, PGE2) and efferent vasoconstriction (mainly Ang II) [78]. ET-1 induced vasoconstriction was also found to be significantly suppressed after its administration [79].
Furthermore, PIO administration blunted the vasoactive responses of Ang II and other adrenergic agonists to the renal vasculature in diabetic spontaneously hypertensive rats; higher renal cortical blood perfusion was observed versus controls [80]. Its activity to modify Ang II induced renal haemodynamic responses was associated with downregulation of the number and expression of renal AT1R receptors [81]. Moreover, studies in insulin receptor substrate (IRS)-1 and IRS-2 knockout mice suggested suppressed kidney autoregulatory function at lowered perfusion pressure levels; they proposed the possible involvement of IRSs in renal haemodynamic abnormalities found in diabetes [82, 83]. This phenomenon was more conspicuous in the deep renal layer compared to the superficial layer. PIO effectively improved those IRS-mediated kidney defects. The main results of major in vitro and preclinical studies that explored the possible improvement of hyperfiltration and intraglomerular pressure after PIO administration are shown in Table I.
Table I
Reduction of hyperfiltration and intraglomerular pressure: Main in vitro and preclinical data of pioglitazone administration during a diabetic state
| Ref. | Year* | Study model (in vitro/preclinical) | Main results |
|---|---|---|---|
| Isshiki et al. [78] | 2000 | Mesangial cells | PIO prevented the evolution of glomerular hyperfiltration through downregulation of the activated DAG-PCK-ERK pathway. This effect was mainly achieved after the suppression of molecules that cause afferent vasodilation (ANP, PGE2) and efferent vasoconstriction (mainly Ang II) |
| Tanimoto et al. [75] | 2004 | KK/Ta mice. A diabetic strain that spontaneously experience T2D, hyperinsulinaemia, dyslipidaemia, mild obesity and proteinuria with early stage of diabetic nephropathy | PIO administration improved hyperfiltration and subsequent glomerular enlargement. The expression of ecNOS was significantly suppressed in the glomerular vessels |
| Asakura et al. [76] | 2012 | 24-week-old male OLETF rats. LET rats were used as nondiabetic controls | PIO administration restored several parameters associated with glomerular hyperfiltration. It improved glomerular surface, kidney weight, creatinine clearance and UACR. All intensities of nNOS and COX-2 in the macula densa were restored to normal levels. Expression levels of desmin and TGF-β were also found to be increased in OLETF rats and were restored after PIO therapy |
| Afzal et al. [80] | 2020 | Diabetic model of hypertensive rats | PIO administration blunted the vasoactive responses of Ang II and other adrenergic agonists to renal vasculature. Higher renal cortical blood perfusion was observed versus controls. PIO and adiponectin were found to have several interactions with RAAS at multiple levels |
| Hashimoto et al. [83] | 2020 | IRS-1 and IRS-2 knockout mice | PIO effectively improved the IRS-mediated kidney defects, which are involved in renal haemodynamic abnormalities found in T2D |
PIO – pioglitazone, T2D – type 2 diabetes, ecNOS – endothelial constitutive NOS, nNOS – neuronal NO synthase, COX-2 – cyclooxygenase 2, OLETF rats – Otsuka-Long-Evans-Tokushima Fatty rats, UACR – urine albumin-creatinine ratio, TGF-β – transforming growth factor β, ANP – atrial natriuretic peptide, PGE2 – prostaglandin E2, Ang II – angiotensin II, DAG-PCK-ERK – diacylglycerol-protein kinase C-extracellular signal-regulated kinase, RAAS – renin-angiotensin-aldosterone system, IRS – insulin receptor substrate; Ref. – Reference;
Renal hypoxia
Almost 10% of total body oxygen consumption is used by the kidneys; 80% from the Na+/K+-ATPase pump in the cortical PCT [6]. Upregulation of SGLT2 during hyperglycaemia leads to enhanced sodium and glucose reabsorption from the PCT; hence it promotes increased ATP consumption and consequently enhanced oxygen utilization from the mitochondrial network [6, 28]. The renal ability to increase renal plasma flow and subsequently its parenchymal oxygen consumption is limited and mainly is correlated with reduced GFR; this in turn promotes a mismatch between oxygen supply and demand and eventually leads to renal hypoxia. Moreover, structural renal abnormalities can promote hypoxic conditions and maintain a vicious self-amplifying process.
Kidney hypoxia is a well-established unifying factor both in early and advanced stages of DKD [6, 28]. It promotes the accumulation of extracellular matrix that destroys normal renal architecture; eventually it promotes fibrosis [84]. Moreover, ample evidence suggests that diabetes can enhance the sensitivity to renal ischemia/reperfusion injury (IRI) through: (i) repression of mitochondrial function and mitophagy; (ii) stimulation of inflammation, oxidative stress, and apoptosis; and (iii) evolution of diffuse tubular necrosis [85, 86].
In vitro and preclinical evidence suggested that PIO administration protected renal IRI through its significant antiapoptotic, anti-inflammatory and antioxidant activities, which were independent of its antihyperglycaemic effect. Its anti-inflammatory effect was mainly expressed by inhibition of the nuclear factor-κB (NF-κB) signalling pathway and its related proteins (p-IKK-β and IκB-α). PIO was also found to: (i) suppress intrarenal endoplasmic reticulum stress; (ii) enhance major renal autophagy mechanisms; and (iii) regulate the activity of the adenosine monophosphate activated protein kinase (AMPK)-mammalian target of rapamycin (mTOR) signalling pathway [87–93].
Diabetic podocytopathy
Podocytes are highly specialized cells of the glomerular basement membrane, which have an active role in preventing the passage of plasma proteins to enter the urinary ultrafiltrate [28, 94]. Podocytes constitutively express functional PPAR-γ, which was found to reduce their apoptotic cell death [95]. When exposed to hyperglycaemic conditions they showed defective autophagy and eventually injury and renal damage [94, 95]. Podocyte detachment and destruction have been associated with urine albumin secretion in a diabetic state [6, 28, 95, 96]. Reduced podocyte density and foot process effacement, as well as lipid vacuolization, have been described both in patients with DKD and in individuals with obesity-related nephropathy [28].
Preclinical studies have shown that PIO improved the expression of several renal podocyte number markers (such as Wilms tumour 1 (WT1) and Zonula occludens-1 (ZO-1)). It reversed structural abnormalities of podocytes and eventually decreased the severity of proteinuria in an obese state. PIO administration decreased urinary sediment podocalyxin excretion, preserved glomerular podocalyxin expression and restored normal function of most podocyte structure-related genes. It also suppressed apoptosis, hypertrophy and cell death, and preserved their differentiation [97–101].
When PIO was administered to a group of patients with uncontrolled T2D, urinary sediment podocalyxin excretion was significantly suppressed, compared to that found in the SU arm. Its podocyte-protective capacity was independent of its antihyperglycaemic activity and was partly shown to be the result of suppressed renal inflammation [102]. Reduction of urinary podocytes was also found in T2D patients with microalbuminuria, suggesting a podocyte protective effect in the early stages of DKD [103].
Mitochondrial dysfunction and injury
Both functional and structural integrity of renal mitochondria have a pivotal role in the modulation of potential hypoxic injury and oxidative stress [6, 28]. Mitochondrial dysfunction, fragmentation and remodelling have emerged as important phenomena in patients with DKD [6, 104]. Mitochondrial balance is mainly achieved through constant and rapid activity of fission and fusion, during which sister mitochondria rapidly come together and split [105]. PIO was found to alleviate mitochondrial dysfunction. It modulated the mitochondrial electron transport chain (ETC) and the activity of dynamic proteins; hence it restored ATP production [106]. PIO therapy also suppressed the activated endoplasmic reticulum stress sensor proteins through restoration of the PPAR-γ coactivator 1α (PGC-1α) prion protein signal transduction pathway; in this way it protected against mitochondrial dysfunction [107].
The intrarenal NLRP-3 inflammasome pathway and ET-1 activity
The nucleotide-binding domain, leucine-rich repeat containing protein (NLRP)-3 inflammasome is an intracellular multimeric protein molecule, which is upregulated in diabetes; it promotes the autocatalytic activation of caspase-1, stimulating the proteolytic activation of proinflammatory cytokines (including interleukin (IL)-1β) [6, 108]. Inhibition of the intrarenal NLRP-3 inflammasome was shown to suppress the evolution of diabetic nephropathy [109]. PIO achieved significant suppression of NLRP3 inflammasomes and their downstream effectors (caspase-1, IL-1β, and IL-18) in the glomerular mesangium. This effect was suggested to delay the progression of DKD in a preclinical model of T1D [110].
Apart from its potent vasoconstrictor activity, which promotes a net increase of efferent arterial renal resistance, higher renal ET-1 levels during hyperglycaemia have been associated with mesangial proliferation, matrix accumulation, podocyte defects, inflammation, and fibrosis [70, 79, 111]. PIO administration in a daily dose of 30 mg was found to suppress urinary ET-1 levels in T2D patients with microalbuminuria, an effect that was beyond its anti-hyperglycaemic activity [112].
Renal inflammation
Hyperglycaemia, hypertension, obesity, IR, and dyslipidaemia can stimulate various aberrant inflammatory signalling pathways, such as apoptosis signalling kinase-1 pathway (ASK1), monocyte chemoattractant protein-1 (MCP-1/CCL2), tumour necrosis factor α (TNF-α) and ILs [6, 28]. These in turn activate intracellular signalling cascades (including Janus kinase/signal transducers and activators of transcription (JAK/STAT) and NF-κB) and maintain a state of low-grade inflammation. This abnormal inflammatory status contributes to renal endothelial dysfunction, glomerulosclerosis, tubulointerstitial remodelling and eventually fibrosis [6, 113]. The chronically activated innate immune system (especially macrophage infiltration in the renal interstitium) has been closely related to the progression of renal decline in DKD [114, 115]. The “classically activated” M1 phenotype stimulates the production of inflammatory cytokines that promote renal tissue damage. M2 macrophages exert an anti-inflammatory effect that contributes to renal tissue repair, having in mind the complexity of stimuli that govern this state. Interestingly, in DKD, the number of M1 macrophages was found significantly increased, while M2 macrophages were reduced [6, 8, 116].
In vitro data suggested that PIO suppressed both IL-6 and TNF-α levels, by suppressing the expression of NF-κB in HEK-293 (a renal cell line) during a hyperglycaemic state [117]. It was also demonstrated that hyperglycaemia promoted higher MCP-1 levels and NF-κB activity in mesangial cells that were suppressed after PIO therapy [118]. PIO administration in animal models of DKD suppressed several molecules that are closely related to inflammation (such as TNF-α, MCP-1, NF-κB, intercellular adhesion molecule-1 (ICAM-1) and mitogen-activated protein kinase (MAP kinase)) and inhibited renal macrophage infiltration – effects that were beyond its antihyperglycaemic control. It also normalized adiponectin levels [119, 120].
Compared to glipizide, PIO therapy suppressed IL-6 levels in T2D patients with overt diabetic nephropathy, an activity that was not solely attributed to its glycaemic control [121]. High-sensitivity C-reactive protein (hs-CRP) levels and erythrocyte sedimentation rate were also reduced in diabetic kidney transplant patients, receiving PIO therapy, compared to placebo [122, 123]. Furthermore, recent evidence suggests that PIO administration ameliorated the peri-renal adipose tissue inflammation in prediabetic rats and reversed their structural, functional and renovascular defects [124].
Increased oxidative stress
Enhanced glucose uptake has been reported in many renal cell populations during a diabetic state (PCT, mesangial and glomerular epithelial cells), which are unable to decrease glucose transport rates and subsequently to prevent the evolution of intracellular hyperglycaemia [125]. High intracellular glucose levels promote the production of oxidants and/or ROS. Eventually the oxidation of important macromolecules, which are closely related to the pathogenesis of DKD (including lipids, proteins, carbohydrates, and DNA), is established [6, 28, 125, 126]. In vitro studies in renal cells during hyperglycaemia suggest that PIO can suppress the expression of membrane receptor for AGEs (RAGE) and can restore cellular antioxidants [117]. In a preclinical model of obesity-related kidney disease, PIO therapy promoted higher AMPK and lower NADPH oxidase 4 (NOX-4) levels; it also suppressed the increased oxidative stress observed [127]. PIO was also found to prevent both renal lipid peroxidation products and free-radical levels; it also stimulated ascorbic acid concentrations and restored renal oxidative balance [128, 129].
Renal fibrosis
The major and final cellular lesion that deteriorates renal function is non-reversible fibrosis and scarring. It is the result of the imbalance between antifibrotic and profibrotic molecular pathways, leading to dysregulation of normal pathophysiological mechanisms involved in renal tissue repair [6]. It is manifested as secondary glomerulosclerosis and/or fibrosis in the tubulointerstitial space [6, 130]. It is characterized by the accumulation of laminin, collagen, fibrillary proteins, activated myofibroblasts and inflammatory cells [6, 28, 131]. MMPs are overexpressed and have been associated with tubulointerstitial fibrosis and eGFR decline [28, 132].
The antifibrotic effects of PIO have been well described in several in vitro and in vivo studies. These effects were mainly found to be independent of its antihyperglycaemic activity and were the result of several mechanisms: (i) suppression of genes that encode for TGF-β1, collagen type I, III and IV, and subsequent reduction of intrarenal concentrations of their corresponding proteins; (ii) inhibition of TGF-β1 induced fibronectin expression in glomerular mesangial cells; (iii) reduction of JAK/STAT-3 pathway activity and downregulation of early growth response factor-1 (EGR-1) and activator protein-1 complex (AP-1) components; (iv) normalization of the imbalance between intrarenal MMP/tissue inhibitors of metalloproteinase (TIMP); (v) downregulation of Twist-1 expression, which has a critical role in kidney fibrosis; (vi) attenuation of the decreased glomerular MMP-2 expression and the increased collagen type IV degradation; (vii) reduction of renal plasminogen activator inhibitor-1 (PAI-1) concentrations; and (viii) through the suppression of IR it can decrease the production of TGF-β1 from mesangial and PCT cells [119, 120, 133–144]. The main results of major preclinical studies in which PIO suppressed the increased inflammation (renal and systemic) and oxidative stress, as well as renal fibrosis, are shown in Table II.
Table II
Main preclinical evidence of pioglitazone in the reduction of increased inflammation (renal and systemic) and oxidative stress, as well as renal fibrosis during an obese and/or diabetic state
| Ref. | Year* | Preclinical model | Main results |
|---|---|---|---|
| Dobrian et al. [129] | 2004 | Obese, hypertensive, Sprague-Dawley rats | PIO administration suppressed the increased renal oxidative stress observed both by reducing free-radical production and by increasing NO production/availability |
| Dong et al. [143] | 2004 | Streptozotocin induced diabetic rats | PIO treatment attenuated the reduced glomerular MMP-2 expression and the increased collagen type IV degradation |
| Ohga et al. [120] | 2007 | Five-week-old Sprague-Dawley rats | PIO suppressed renal macrophage infiltration and the expression of TGF-β, ICAM-1 and type IV collagen. It also decreased renal NF-κB activity |
| Ko et al. [119] | 2008 | OLETF rats with T2D | PIO treatment downregulated several inflammatory and profibrotic genes in renal cortical tissues including NF-kB, TGF-β, MCP-1/CCL2, PAI-1 and VEGF. It also suppressed macrophage infiltration and NF-κB activation in association with decreased type IV collagen and TGF-β1 expression |
| Toblli et al. [142] | 2009 | OZR and ZDFR. LZR were used as controls | PIO administration suppressed renal fibrosis, as shown by the improvement of glomerulosclerosis, tubulointerstitial fibrosis, tubular atrophy and podocyte injury indexes. It was also demonstrated that PIO exerted significant anti-oxidative and anti-inflammatory activities |
| Toblli et al. [144] | 2011 | ZDFR and LZR | PIO administration normalized the renal levels of fibronectin, connective tissue growth factor, TNF-α, IL-6, VEGF and MCP-1 |
| Wang et al. [110] | 2017 | ApoE (-/-) diabetic mice | PIO achieved significant suppression of NLRP3 inflammasomes and their downstream effectors (caspase-1, IL-1β, and IL-18) in the glomerular mesangium. This effect was suggested to delay the progression of DKD in this preclinical model |
| Li et al. [127] | 2019 | Ob/ob mice and homologous C57BL/6 (wild-type) mice | PIO therapy promoted higher AMPK and lower NOX-4 levels; it also suppressed the increased oxidative stress observed |
| Wang et al. [137] | 2019 | Male ZDF rats | PIO administration downregulated Twist-1 expression, which has a critical role in renal fibrosis. This activity was independent of its glycaemic effect |
PIO – pioglitazone, T2D – type 2 diabetes, DKD – diabetic kidney disease, TGF-β – transforming growth factor β, MCP-1/CCL2 – monocyte chemoattractant protein-1, NF-κB – nuclear factor-κB, TNF-α – tumour necrosis factor α, VEGF – vascular endothelial growth factor, PAI-1 – Plasminogen activator inhibitor type 1, ICAM-1 – intercellular adhesion molecule-1, apoE – apolipoprotein E, IL – interleukin, NLRP3 – nucleotide-binding domain, leucine-rich repeat containing protein, NOX-4 – NADPH oxidase 4, AMPK – 5′ AMP-activated protein kinase, MMP-2 – matrix metalloproteinase-2, NO – nitric oxide, OLETF rats – Otsuka-Long-Evans-Tokushima Fatty rats, OZR – obese Zucker fa/fa rats, ZDFR – ZDF fa/fa rats, LZR – lean Zucker rats, ZDF rats – Zucker diabetic fatty rats, Ref. – Reference;
Other possible mechanisms of kidney protection
Other possible nephroprotective mechanisms of PIO in a diabetic state are: (i) normalization of renal cell cycle homeostasis and angiogenesis though regulation of vascular endothelial growth factor activity; (ii) stimulation of renal endogenous stem cells; (iii) increased tubular cell albumin uptake; (iv) significant improvements of contractile dysfunction in renal mesangial cells during hyperglycaemia; (v) normalization of renal klotho expression after inhibition of Ang II activity; and (vi) renal lipotoxicity is an important contributing factor to the development of DKD – PIO suppressed the increased renal lipid content and its lipotoxic damage [68, 145–150]. A phase II study has also been also recently launched and is recruiting individuals, to investigate the possible activity of PIO in suppressing muscle sympathetic nerve overactivity in patients with CKD [151].
Combination of PIO with RAAS blockers
Evidence from preclinical models of obesity, metabolic syndrome and T2D has demonstrated that the combination of PIO with Ang II receptor blockers (ARBs) was more effective compared to either drug alone in ameliorating the progression of the existing glomerulosclerosis. This combination was shown to have multiple beneficial effects on systolic BP, proteinuria, interstitial fibrosis, the glomerulosclerosis index and eGFR decline. It also reduced PAI-1 levels, suppressed macrophage/monocyte infiltration, preserved glomerular podocyte function and stimulated AT2R and Mas expression. Suppression of renal oxidative stress and of inflammation were also other possible additive effects of this combination [152–157]. Indeed, small, short-term clinical studies in patients with DKD have shown that the combination of PIO with ARBs achieved significant additive effects in preserving serum creatinine levels, eGFR and reduced UACR [158, 159].
Combination of PIO with SGLT-2 inhibitors
Encouraging results were obtained in a preclinical study that investigated the possible combination of PIO and dapagliflozin (DAPA) to prevent DKD [160]. Specifically, 8-week-old male db/db mice were divided into four arms: (i) vehicle arm (n = 5); (ii) PIO in a daily dose of 30 mg/kg (n = 8); (iii) DAPA in a dose of 2 mg/kg/day (n = 8); and (iv) combination of PIO and DAPA in the above doses (n = 7). Both DAPA and PIO monotherapy reduced glomerular hypertrophy, while the combination therapy arm achieved the greatest reduction in terms of both glomerular tuft area and mesangial expansion ratio. Furthermore, the lowest number of apoptotic renal cells was found in the combination arm. Mice treated with the combination arm showed the best preservation of glomerular morphology and the most well-preserved glomerular structures. Since there was no statistically significant difference in blood glucose levels between the three treatment groups, it was suggested that the combination of PIO with DAPA exerted renoprotective mechanisms beyond their individual glucose lowering effects. A randomized phase II study is recruiting patients and is going to investigate the possible role of a triple combination therapy with PIO, a SGLT2 inhibitor and MET versus MET with or without the combination with other oral antihyperglycaemic medications (DPP-4 inhibitors, SUs and acarbose), to decrease hsCRP in T2D patients with a history of ischaemic stroke [161]. A secondary endpoint of this study is the estimation UACR in active versus control arms.
Combination of PIO with GLP-1R agonists
Early intensive triple therapy with MET, PIO and exenatide was shown to produce greater and more durable A1C reductions in patients with new-onset T2D compared to sequential add-on therapy with MET followed by SU and then basal insulin; patients treated with triple combination therapy experienced less hepatic steatosis and fibrosis compared to those in the conventional therapy group [162, 163]. Improvement of painful diabetic neuropathy and corneal nerve regeneration were also demonstrated, while the generalizability of these results in other diabetic microvascular complications is anticipated [164].
Conclusions
The implication of multiple metabolic, haemodynamic, inflammatory, and tubular pathways in the pathophysiology of DKD generates the need for multitargeted treatment approaches, to improve the disease evolution at all levels and delay or even reverse its progression [6, 28, 165–167]. It has been shown that IR is an independent predictor of CKD and of its cardiovascular and renal complications [8, 11, 168, 169]. PIO has a unique mechanism of action and several important pleiotropic effects. It has shown significant cardiovascular benefits, improvements of several components of the metabolic syndrome and beneficial effects on non-alcoholic fatty liver disease (NAFLD)/non-alcoholic steatohepatitis (NASH) [11, 22, 170]. Its possible role in other IR states is also under intense investigation, both as monotherapy and in combination with other agents [11, 22, 171].
Preclinical evidence, small, short-term clinical studies and meta-analyses of these studies have consistently shown that PIO may have nephroprotective effects beyond its antihyperglycaemic activity. Animal studies have also shown that it can act beneficially on several fundamental cellular pathways involved in the pathogenesis and progression of DKD (Figure 1). These effects can possibly underly its ability to slow the progression of DKD. PIO could also have additive effects, when combined with RAAS inhibitors and SGLT-2 inhibitors. Experimental evidence is ongoing to investigate and confirm all these possible beneficial mechanisms. The main results of prospective randomized clinical studies that explored the possible role of PIO in DKD are shown in Table III.
Table III
Pioglitazone and kidney protection: main results of prospective randomized clinical studies during a diabetic state
| Ref. | Year* | Study population | Main results |
|---|---|---|---|
| Nakamura et al. [112] | 2000 | Forty-five NIDDM patients with microalbuminuria were randomized to three arms: (i) PIO arm (n = 15); (ii) GLIBE arm (n = 15); and (iii) VOGLI arm (n = 15). Patients were treated for 3 months | Only PIO administration was found to suppress UACR and urinary ET-1 levels. This effect was beyond its anti-hyperglycaemic activity |
| Nakamura et al. [103] | 2001 | Twenty-eight normotensive T2D patients with microalbuminuria and 30 age-matched normotensive controls were enrolled. T2D patients were randomized to two arms: (i) PIO (n = 14); and (ii) placebo (n = 14). Treatment duration was 6 months | PIO administration reduced both UACR and the number of urinary podocytes. A possible podocyte protective effect of PIO in the early stages of DKD was suggested |
| Agarwal et al. [121] | 2006 | Forty-four patients with T2D and overt diabetic nephropathy were randomized to receive either PIO (n = 22) or GLIPI (n = 22) for a total of 16 weeks | Compared to GLIPI, PIO therapy suppressed IL-6 levels and WBC, activity that was not solely attributed to its glycaemic effect |
| Jin et al. [158] | 2007 | Sixty T2D patients with stage 3 or 4 DKD were randomized to receive either PIO and LOSA (n = 30) or LOSA monotherapy (n = 30) | The combination of PIO/LOSA resulted in significantly lower serum creatinine levels at 12 months and in significantly lower proteinuria levels at 6 and 12 months. The declines in creatinine clearance and GFR rate below baseline measurements were significantly slower in the combination arm versus the monotherapy arm |
| Schneider et al. [18] | 2008 | Post hoc analysis from the PROactive study that investigated the relationship between CKD and incident CVD | 467 (20.4%) patients in the PIO arm experienced CKD versus 370 (16.3%) patients in the placebo arm (p = 0.0004) |
| Morikawa et al. [159] | 2011 | Patients with T2D and microalbuminuria were randomized to receive either PIO (n = 32) or MET (n = 31) for 52 weeks. All patients were treated with RAAS inhibitors | Changes in the log-UACR from baseline were –8.3% in the PIO arm and +4.2% in the MET arm (p = 0.01), despite similar BP and glycaemic changes |
| Xing et al. [102] | 2012 | Ninety-eight patients with uncontrolled T2D were randomized to receive either add-on therapy with PIO (n = 49) or SU (n = 49) for 12 weeks | The urinary sediment podocalyxin excretion in the PIO arm was significantly decreased, compared to that found in the SU arm. The podocyte-protective capacity found after PIO administration was independent of its antihyperglycaemic activity and was partly shown to be the result of suppressed renal inflammation |
| Kharazmkia et al. [122] | 2014 | 62 patients with diabetes and kidney transplantation were randomized to receive either PIO or placebo for a total of 4 months | hs-CRP levels and ESR were reduced in diabetic kidney transplant patients receiving PIO therapy, compared to placebo |
| Arashnia et al. [123] | 2015 | 58 diabetic patients, who had undergone renal transplantation, were randomized to two arms: (i) PIO in combination with insulin; and (ii) insulin with placebo | hs-CRP was reduced by 4.82 mg/dl in the PIO arm versus 1.93 mg/dl in the placebo arm (p = 0.007) |
PIO – pioglitazone, SU – sulfonylurea, GLIBE – glibenclamide, VOGLI – voglibose, GLIPI – glipizide, LOSA – losartan, RAAS – renin-angiotensin-aldosterone system, MET – metformin, T2D – type 2 diabetes, NIDDM – non-insulin dependent diabetes, DKD – diabetic kidney disease, CKD – chronic kidney disease, CVD – cardiovascular disease, UACR – urine albumin-creatinine ratio, ET-1 – endothilin-1, IL – interleukin, WBC – white blood count, hs-CRP – high sensitivity C-reactive protein, ESR – erythrocyte sedimentation rate, GFR – Glomerular filtration rate, BP – blood pressure, PROactive – PROspective pioglitAzone Clinical Trial In macroVascular Events; Ref. – Reference;
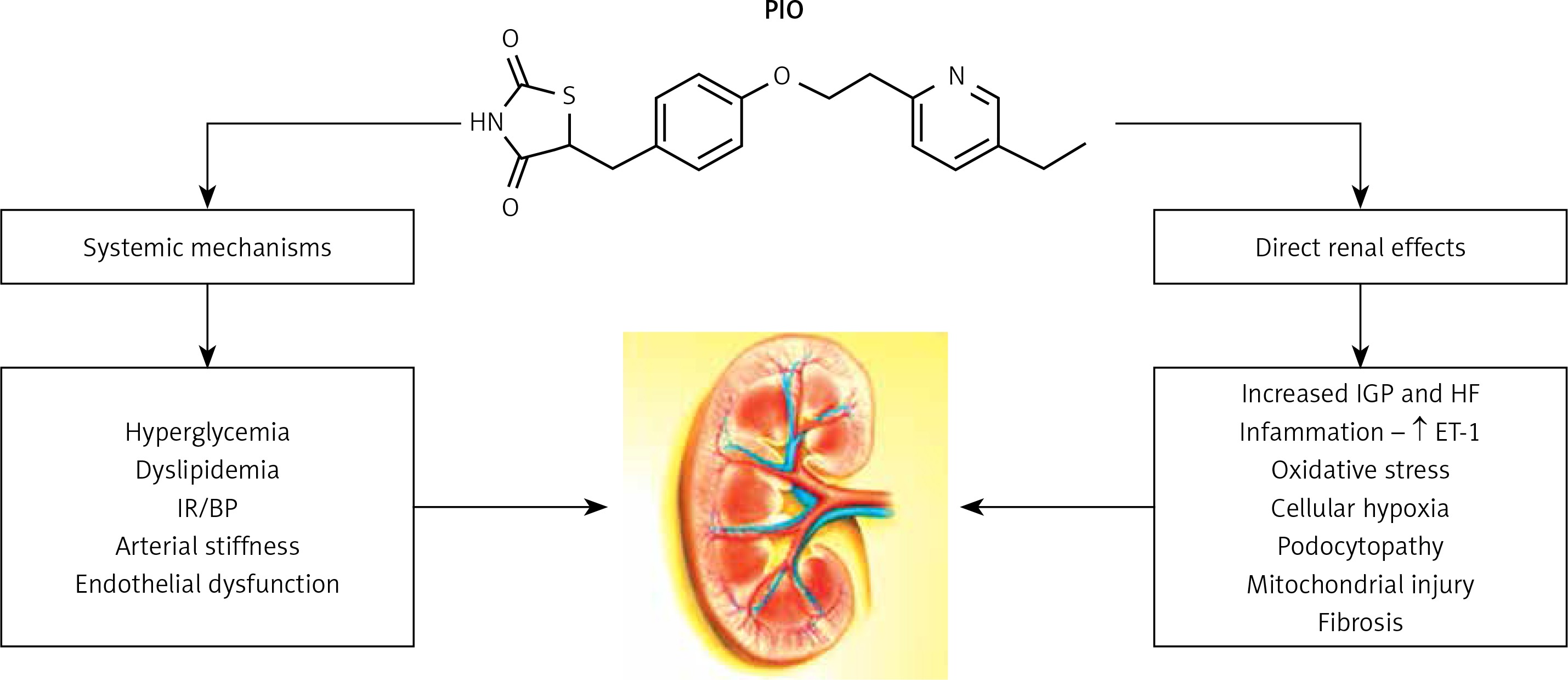
Figure 1
Pioglitazone in diabetic kidney disease: possible mechanisms of renoprotection
PIO – pioglitazone, IR – insulin resistance, BP – blood pressure, ET-1 – endothelin-1, IGP – intraglomerular pressure, HF – hyperfiltration.
Fluid retention and oedema are the most common adverse effects of TZD therapy (approximately 7% of patients treated in monotherapy and 15% after combination with insulin); it is more pronounced in patients with CKD [8, 11, 22]. This is mainly the result of increased fluid reabsorption in the distal nephron, as well as increased vascular permeability in adipose tissues. It could be easily controlled after the addition of a distally acting diuretic to the therapeutic strategy [172]. However, low-dose of PIO therapy (7.5–15 mg) can be significantly effective and more tolerable with less adverse effects on oedema, weight gain and heart failure, although this approach should be further explored [173, 174]. Interestingly, in the Thiazolidinediones Or Sulfonylureas Cardiovascular Accidents Intervention Trial (TOSCA.IT) study no difference for oedema and rates of heart failure were found between PIO (mean daily dose: 23 mg) and SU arms [175]. Furthermore, the osmotic diuresis induced by SGLT2 inhibitors has been found to promote mainly reduction of the interstitial volume rather than the intravascular volume. This effect can be particularly helpful in the suppression of PIO-induced sodium and fluid retention [22, 176, 177].
It is of vital importance to investigate the effectiveness of PIO, both as monotherapy and its combinations with SGLT2 inhibitors and/or GLP-1R agonists in randomized controlled trials (RCTs), investigating “hard” renal endpoints (such as eGFR decline and/or slope, initiation of renal replacement therapy and renal associated death) in different renal phenotypes [6, 178]. Since multiple medications in combination will be needed to tackle DKD, PIO may complement these different drug categories in consonance, seemingly by entirely different or complementary pathophysiological mechanisms [6, 28]. In the meantime, results from future well-designed observational studies in a real-life setting can give valuable information for the possible beneficial effects of PIO and its therapeutic combinations in terms of kidney protection. Any possible safety signals should be thoroughly monitored and appropriately evaluated.