Introduction
Dyslipidemia is a frequently seen disorder, and its control is necessary for atherosclerotic prevention worldwide [1–5]. Animal and genetic studies have led to an improved pathophysiological understanding of lipid disorders and the development of lipid-modifying therapies [6–10]. Understanding the molecular basis of animal models with dyslipidemia is thus useful for future research [7–9].
A mouse strain, derived from an inbred strain of Japanese wild (original)-type mice (KOR; Mus musculus molossinus) was established, showing high plasma cholesterol concentrations with severe cutaneous xanthomatous lesions. This mouse strain was termed spontaneously hyperlipidemic (SHL) mice [11]. The phenotypes of SHL mice are known to be transmitted in an autosomal recessive manner [11–13]. Given the spontaneously derived nature, SHL mice are not regulated by the basic rule of the Cartagena Protocol on Biosafety [14], although such regulations are generally applied to transgenic animals. Thus, SHL mice have advantages with regard to their utility for research.
As one of the most important genes related to lipid metabolism is the apolipoprotein E (Apoe) gene, Apoe-deficient mice, transgenic animals created by homologous recombination in embryonic stem cells, exhibit severe hypercholesterolemia and atherosclerosis. These mice are used in research on lipid disorders [15–17]. Of note, SHL mice also exhibit disruption of the Apoe gene and the absence of APOE protein, with severe hyperlipidemia and atherosclerotic manifestations [11–13].
The disruption of the Apoe gene is suggested to be associated with the development of hyperlipidemia in SHL mice [12], so it is important to understand the workings of the Apoe gene; however, no detailed description of the Apoe gene in SHL mice has yet been obtained. The present study therefore determined the complete DNA sequences of the whole Apoe gene of SHL mice, which are expected to prove useful for future research on lipid disorders based on the molecular basis of SHL mice.
Material and methods
Animals
The present study compared SHL mice to wild-type KOR mice as normal controls. The study was reviewed and approved by the Animal Care and Use Committee of Animal Ethics of the University (No. 29K-36).
Reverse transcription polymerase chain reaction (RT-PCR) and PCR procedures
Genomic DNA and complementary DNA (cDNA) analyses were conducted via the usual methods in the present study. Genomic DNA was extracted from mouse liver using a DNeasy Blood & Tissue kit (Qiagen Science, Germantown, MD, USA). PCR amplification was performed using an Accuprime Taq DNA Polymerase System (Thermo Fisher Scientific, Inc., Waltham, MA, USA) in a 10-μL reaction volume containing 10 mM Tris-HCl (pH 8.4), 1.5 mM MgCl2, 50 mM KCl, 0.2 mM each of the dNTPs, 0.5 mM of each primer, and Accuprime Taq DNA polymerase. For RT-PCR, the total RNA sample was extracted from the mouse spleen using an RNA extraction kit (Qiagen Science) as described in the manufacturer’s protocol. Following DNaseI treatment, total RNA was reverse-transcribed into cDNA using Superscript III reverse transcriptase with Oligo(dT)12-18 primers (Thermo Fisher Scientific, Inc.). After the initial denaturation for 2 min at 94°C, a 3-step PCR program was carried out as follows: 35 cycles of 20 s at 94°C for denaturation, 30 s at the respective primer-specific annealing temperature, and 40 s at 68°C for extension. The PCR program was followed by a final extension period of 7 min at 68°C and a cooling phase at 4°C. The amplified PCR products were separated by electrophoresis on an agarose gel followed by staining with ethidium bromide and detection by UV transillumination.
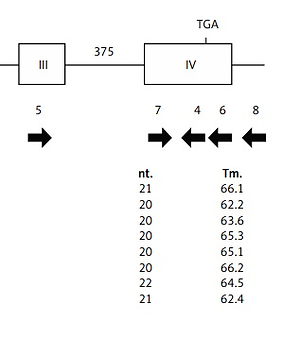
The disruption of the Apoe gene of SHL mice was evaluated by comparing the PCR products. Using several primer sets spanning each entire exon or intron (Figure 1), mutation regions were examined by comparing the length of PCR products between SHL and control KOR mice.
DNA sequencing
PCR amplification was carried out with several primers spanning almost the entire exon and intron regions at the first step of DNA sequencing of the whole Apoe gene. After confirming the amplification of PCR products, the resulting fragments were applied to direct DNA sequencing. The PCR products for insertion/deletion in the Apoe gene were cloned into a plasmid vector using the PCR4-TOPO-TA cloning kit (Thermo Fisher Scientific, Inc.). Nucleotide sequences were determined by Sanger sequencing. The mutation site and cDNA structures were then examined with the DNA sequences.
Results
PCR amplification using some pairs of primers covering each exon and intron of the Apoe gene of the control KOR mice detected the mutation site in the Apoe gene of SHL mice. PCR products specific to SHL mice were observed, with DNA sequences revealing a 4700-bp insertion located between the 7th and 8th nucleotides from the 5′ end of exon 4 of the Apoe gene of SHL mice, dividing exon 4 into two regions (Figure 2). The insertion contained two 365-bp long terminal repeats (LTRs) with the same direction and a 2730-bp transposal element. In addition, both ends were flanked by a 6-bp target site duplication (TSD) of the sequence ATGGAG. No products specific to SHL were observed in the intron 3 regions (data not shown).
Figure 2
Schematic figure of the mutation site structure in the Apoe gene of SHL mice. The insertion occurred between the 7th and 8th nucleotides from the 5′ end in exon 4 of the Apoe gene, which divided exon 4 into two regions. The insertion was 4700 bp in length, and it contained two long terminal repeats (LTRs) of 365 bp at each terminal. Both ends were flanked by a 6-bp target site duplication (TSD) of the DNA sequence ATGGAG

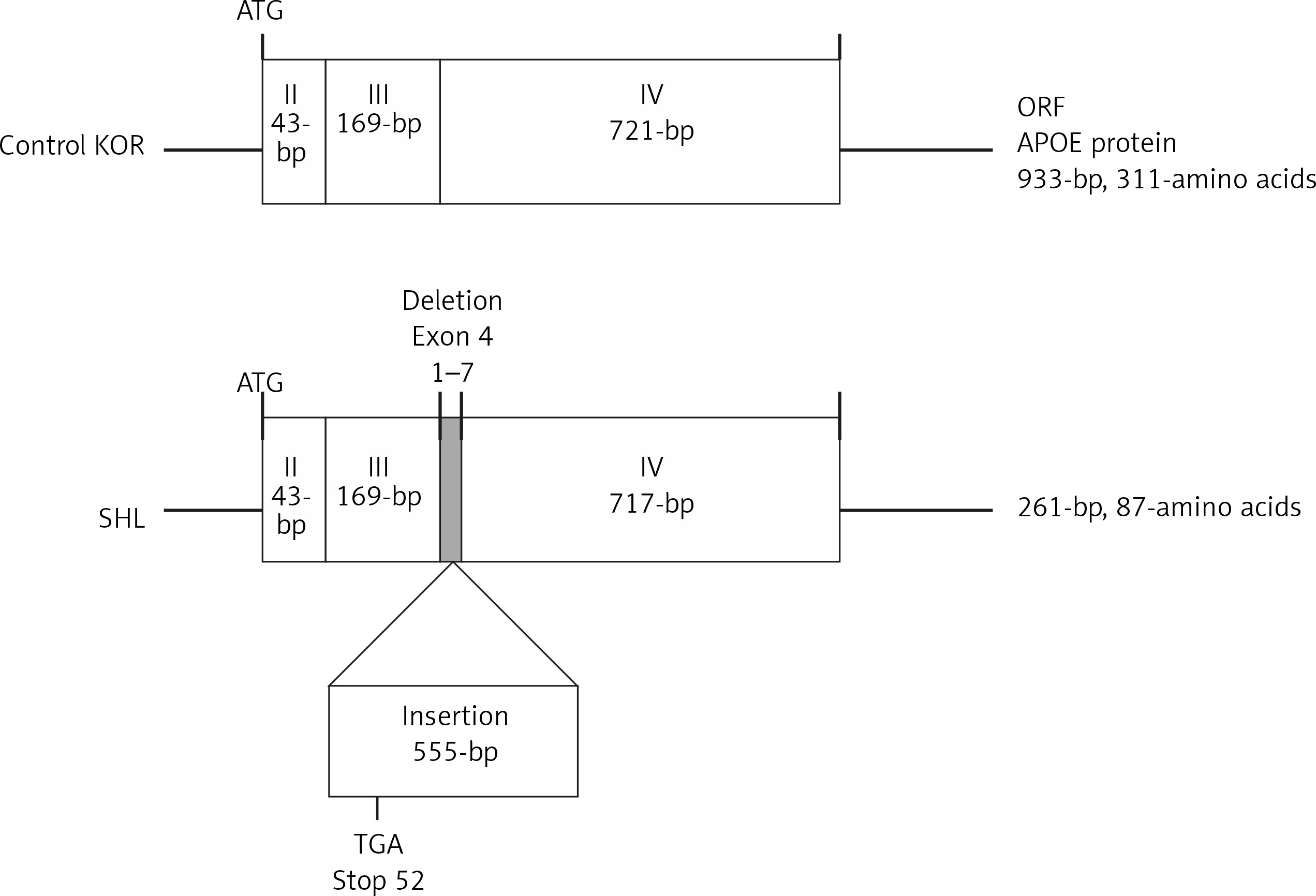
On RT-PCR using primers spanning the whole Apoe coding region from the starting codon on exon 2 to the stop codon on exon 4, the PCR product for the Apoe cDNA, including the open reading frame (ORF), appeared to be 0.5 kb longer than the product of the control KOR mice (data not shown). Direct sequencing showed that the Apoe gene of SHL mice differed from that of control KOR mice, containing a 555-bp insertion between the 3′ end of exon 3 and the 5′ end of exon 4 in SHL mice (Figure 3). In addition, there was a deletion of a 7-bp sequence (the 1st to 7th nucleotides) from the 5′ region of exon 4. The inserted fragment of SHL mice was able to induce frameshift by an early stop codon (TGA), resulting in a protein product that consisted of 87 amino acids in SHL mice compared to one of 311 amino acids in control KOR mice.
Figure 3
Schematic figure showing a comparison of the cDNA structures estimated from RT-PCR amplification and direct sequencing between SHL and control KOR mice. Regarding the differences in the cDNA sequences, one was a deletion of 7 nucleotides (1st to 7th nucleotides) in the 5′ end of exon 4 in the Apoe gene, and another was a 555-bp insertion between the 3′ end of exon 3 and the 5′ end of exon 4 in the Apoe gene. The frameshift induced by an early stop codon (TGA) in the insertion can affect the amino acid sequence in the APOE protein in SHL mice. The APOE protein in normal mice was estimated to contain 311 amino acids in control KOR mice but only 87 amino acids in SHL mice
Discussion
The present study clarified the molecular basis of the Apoe gene of SHL mice in comparison to that of wild-type KOR mice as controls. In SHL mice, the insertion divided exon 4 of the Apoe gene into two regions. The center region of the insertion was 4700 bp in length with 2 DNA fragments of LTR 365 bp in size located at both terminal sites, flanked by terminal 6-bp repeats. In the 555-bp insertion and the 7-bp deletion found in the cDNA structure, the inserted fragment could induce a frameshift of an early stop codon with an altered acceptor site for mRNA splicing, thereby leading to disruption of the Apoe gene and protein in SHL mice (Figure 4). The findings of the present study are expected to aid in future research on lipid disorders using SHL mice.
Figure 4
Schematic figures showing the splice sites as estimated from comparisons of the genomic DNA sequences and cDNA structures between SHL and control KOR mice. The inserted fragment changes the acceptor site for mRNA splicing. This produces a frameshift due to an early stop codon (see the schema and footnote of Figure 3)

The insertion in exon 4 is thought to be an important determinant of characteristics of SHL mice. While the exact reasons the phenomenon spontaneously occurred remain unclear, we offer some evolutionary insights into the mutated disorders, as previously suggested [11, 18, 19]. The insertion seemed to be a ‘retrotransposon’ element with two 365-bp LTRs and two 6-bp TSDs. In using an in silico estimation (Hitachi Solutions, Ltd., Dnasis Pro, Tokyo, Japan), the ORFs included the longest mRNA, composed of 2730 bp coding 909 amino acids in the transposal element and 195 bp coding 64 amino acids in the LTR region. In a BLAST protein analysis (Standard Protein BLAST: https://blast.ncbi.nlm.nih.gov/Blast.cgi?PROGRAM=blastp&PAGE_TYPE=BlastSearch&LINK_LOC=blasthome) to predict the origins including the inserted DNA fragment, several proteins related to LTR retrotransposons, e.g. Gag (encoding group-specific retroviral antigen), Pol (encoding the reverse transcriptase/ribonuclease H) and integrase, were detected. The inserted fragment coding proteins strongly indicated infection with LTR transposons. In typical LTR retrotransposons, endogenous retrovirus sequences (ERVs) contain 2 LTRs (ranging from 300 to 1000 bp) and fragments (around 6 to 9 kb) encoding the viral Gag, Pol and Env (envelope) proteins between LTRs and are flanked by a short duplication (4 to 6 bp) of host sequences. Close to 40% of the mouse genome is made up of fossils of transposable elements, of which approximately 10% represent ERVs [19, 20]. Thus, the insertion in SHL mice may have originated from an ERV infection.
The present findings implicate one possible mechanism for the sudden occurrence of lipid disorder. In addition, the Apoe gene and APOE protein play critical roles in lipid metabolism and atherogenesis (in fact, an increase in very low-density lipoprotein and remnant and a decrease in high-density lipoprotein are seen in APOE deficiency), and some Apoe-modulating models (e.g. Apoe knockout mice) have been developed [9, 11]. By comparing the lipid profiles between those models and SHL mice with known molecular backgrounds, the elucidation of lipid disorders and atherogenic mechanisms can further progress.
In conclusion, in the present study, we found an insertion of exon 4 of the Apoe gene in SHL mice, and a frameshift of an early stop codon induced by the mutation could lead to disruption of the Apoe gene. The insertion may be a retrotransposon element. The findings are expected to prove useful for future research on lipid disorders based on the molecular basis of SHL mice.